Innovative technology at highly competitive prices.
|
|
ELIT Brand Electrochemical Sensors and Computer-based Instrumentation. Innovative technology at highly competitive prices. |
| HOME | PRODUCTS | TECHNICAL INFORMATION | SITE MAP | CONTACT US | PRICE LIST |
How Ion-Selective Electrodes Work Click here for a printer-friendly PDF version Click here for examples of possible arrangements of ISE's in practical applications. Some Basics There are two different types of Electrical Conductivity: 1) In Metals the electric current is carried by Electrons. 2) In Liquids the electric current is carried by Ions Every Electrochemical Process (Galvanic Cell, Electrolysis, Electro-Analysis) involves both these types of conductivity. The junctions where they meet and transfer the electrical charge are referred to as Metal-Liquid Interfaces. These interfaces were originally called Electrodes, but now this term is also used for various other devices such as welding electrodes or electro-cardiogram electrodes. At the Metal-Liquid interface there is an exchange of Electrons in one or other direction (details can be found in standard chemistry text books, in sections on Galvanic or Electrolytic Cells. (NB: Galvanic [Voltaic] Cells generate electricity; Electrolytic Cells consume electricity). For example, in a Copper-Silver Galvanic Cell, on one electrode an Oxidation reaction takes place: Cu (metallic) à Cu 2+ (ionic, in solution) +2 e- on the other electrode a Reduction reaction takes place: Ag+ (ionic, from solution) + e- à Ag (metallic - deposited on electrode surface) This explains how the electric current in the wire (Electrons) becomes a current in the liquid (Ions).
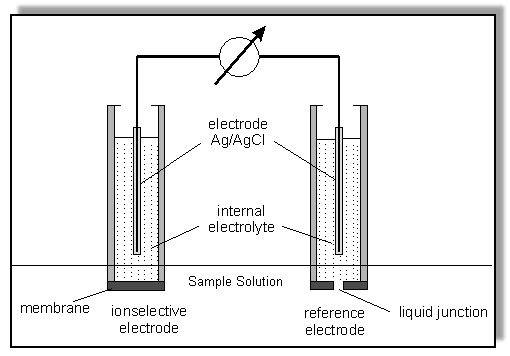
The Electrochemical Circuit for an Ion Selective Electrode measurement. An ISE (with its own internal reference electrode - more details later) is immersed in an aqueous solution containing the ions to be measured, together with a separate, external reference electrode. (NB: this external reference can be completely separate or incorporated in the body of the ISE to form a Combination Electrode.) The electrochemical circuit is completed by connecting the electrodes to a sensitive milli-volt meter using special low-noise cables and connectors. A potential difference is developed across the ISE membrane when the target ions diffuse through from the high concentration side to the lower concentration side (a detailed description follows later).
Figure adapted from that by Wojciech Wroblewski at CSRG, University of Warsaw, Poland. (Copied from http://www.ch.pw.edu.pl/~dybko/csrg/tutorials/ise/index.html) General principle of ISE analysis
At equilibrium, the membrane potential is mainly dependent on the concentration of the target ion outside the membrane and is described by the Nernst equation. Briefly, the measured voltage is proportional to the Logarithm of the Activity (effective concentration) of the ions in solution.
The membrane potential cannot be measured directly. It needs a Metal-Liquid interface (or a metal-solid solution interface in modern "all-solid-state" ISEs) on both sides of the membrane. Theoretically these could just be metal wires immersed in the solutions. But the electrical potential on many simple metal-liquid junctions is not stable; thus the need for a so-called reference system on both sides of the ISE membrane, with a particular metal-liquid interface which is known to have a stable potential. The magnitude of this potential need not be known because it is the same for all measurements of standards and samples and is thus eliminated during the calibration process. Nevertheless, it must be noted that this potential, plus any others that may be generated at any or all of the metal-liquid or liquid-liquid junctions in the circuit, is the value which is seen when the electrodes are immersed in pure water or any other solution which does not contain the target ion. This explains why the measured voltage is not expected to be zero when no target ion is present and also why it is not necessarily always positive when the target ion is present - it all depends on the difference between the ISE voltage and the sum of all the other voltages in the circuit. For example, for a monovalent positive ion, the voltage could be -25 mV in 10ppm and +30mV in 100ppm (or even -60 mV in 10ppm and -5mV in 100ppm) but this still gives a slope of +55mV per decade of concentration and indicates that the ISE is functioning correctly. Reversing the charges above would describe the situation for a monovalent negative ion. It should be noted here that immersion in pure water should be avoided because it tends to leach out the target ion from the ISE membrane. This, together with the inherent instability of the liquid junction potential of the reference electrode, will cause an unstable voltage to be measured in pure water and require the ISE membrane to be re-equilibrated in a high concentration "pre-conditioning" solution before it will give stable readings again. In practice, the most common reference system is a silver wire coated with solid silver chloride and immersed in a concentrated solution (known as the "filling solution") of potassium chloride saturated with silver chloride. The reference electrode is a half-cell that provides a constant potential which is dependent only on the concentration of chloride ions in the filling solution. The reversible Redox reaction involves the chloride atoms in the solid silver chloride (plated on the silver wire) receiving an electron and the chloride ion going into solution, and vice versa. This electrode will give a constant potential of +205 mV (relative to the Standard Hydrogen Electrode) with a saturated KCl/AgCl solution at 25°C.
Electrochemical Processes in the Membrane of an ISE There are various different charge-transfer processes, both outside and inside the membrane for the various different membrane types, and many of these are highly complex and poorly understood in detail. For example, even the apparently simple glass membrane of a pH electrode, which has traditionally been thought of as involving the passage of Hydrogen (H+), or possibly hydroxonium (H3O+) ions, has recently been shown, by radioactive tagging experiments, to involve only the movement of Sodium (Na+) ions ! The following descriptions of the Calcium and Fluoride ISEs are typical examples of the basic principles of ion-selective membrane processes. Nevertheless, it must be noted that these processes may be far more complex than those described and may involve several layers of ions at each phase junction.
Principle of Operation of the Calcium ISE A Calcium ISE has a PVC membrane which is impregnated with an organic molecule which selectively binds and transports Ca2+ ions, and contains an internal solution with a fixed concentration of calcium chloride - added to the KCl / AgCl solution of the internal reference system. (Note that in modern all-solid-state ISEs the internal "solution" is in a solid form). Initially when this electrode is immersed in a sample solution containing Ca2+ ions, the potential difference across the membrane is zero - because there are, on both sides of the membrane, solutions where the electrical charges are balanced (i.e. they contain equal numbers of cations and anions). But very soon after immersion, calcium ions will start to diffuse across the membrane, from the side with the higher calcium concentration to the side with the lower calcium concentration. (NB: for the purposes of this explanation it is convenient to assume that the flow of ions is from the test solution into the ISE.) As the positive calcium ions are transported across the membrane by the diffusion pressure, there is a build up of positive charge (cations) on the inside of the membrane and a corresponding increase in negative charge (anions) outside. These charges on the membrane surface mean that an electrical potential difference is established across the membrane. This potential difference causes the calcium ion migration to slow down and finally stop when the diffusion pressure due to the difference in concentration is exactly balanced by the electric field effects due to the fact that similarly charged particles repel one another. The potential difference at equilibrium is called the membrane potential. Inside the ISE, the build up of positive charge at the membrane surface causes silver ions in the internal reference system to lose their charge (by receiving electrons from the silver wire) and be deposited on the wire. Thus electrons are drawn through the external wiring from the meter and thence from the external reference electrode. Here, chloride ions are attracted to the silver chloride-coated wire and give up their electrons by combining with silver atoms in the wire, and potassium ions flow out into the sample solution through the porous frit (labelled liquid junction in the diagram) to compensate for the positive charge deficiency caused by the loss of calcium. At equilibrium, the electron flow ceases, i.e. there is no current, - but there are residual voltage differences at each metal-liquid, solid-liquid, solid-solid and liquid-liquid junction - in addition to the membrane potential and the reference electrode stable voltage. The measured potential difference, (in millivolts) is the sum of all these potentials. In theory, during calibration with standard solutions of known concentration, and during sample measurement, only the membrane potential is changed so that the other voltages can be ignored. In practice some of these vary a little - particularly at the porous frit (liquid junction) of the reference electrode - and are one of the sources of error in ISE measurements.
Principle of Operation of the Fluoride ISE. In the case of the F ISE, the ion-selective membrane is a single crystal of Lanthanum Fluoride (LaF3) doped with Europium Fluoride (EuF2) which produces holes in the crystal lattice through which F ions can pass. When immersed in a fluoride solution and connected via a voltmeter to an AgCl/KCl external reference electrode immersed in the same solution, the negative F ions in the solution pass through the crystal membrane by normal diffusion from high concentration to low concentration until there is an equilibrium between the force of diffusion and the reverse electrostatic force due to repulsion between particles of similar charge. On the other side of the membrane there is a corresponding build-up of positive ions. The build up of negative F ions on the inside of the membrane is compensated for by Cl ions in the internal reference solution becoming neutralised by combining with the Ag/AgCl wire, and electrons are thus forced through the external wire to the voltage measuring device (ion meter or computer interface). The other terminal of the voltmeter is connected to the Ag/AgCl wire of the external reference electrode. Here, the influx of electrons causes Ag ions in the filling solution to accept electrons and deposit on the silver wire and, consequently, Cl ions to flow out into the sample solution. Note that, in general, depending on the concentrations inside and outside the membrane and which ion is being measured, all the reactions described above could occur in the opposite direction.
Useful References containing recent research: David C. Harris (2001) Exploring Chemical Analysis, 2nd Ed. ISBN 0716735407 Skoog, West, Haller & Crouch (2000), Analytical Chemistry, 7th Ed. ISBN 0030202930 Garry D. Christian (1994), Analytical Chemistry. ISBN 0471305820
CCR/HK/Nico2000Ltd./Sept. 2004 - Last Update 19 Dec. 2014 |