 |
Back to Ion Analysers page | Back to Electrodes page
|
Back to Ion Analysers page | Back to Electrodes page
Standard
Addition & Sample Addition methods for Ion Selective Electrodes.
(only available on ELIT ISE/pH 2-channel Ion Analysers)
A full description of these methods is given below this example of a typical screen display from the ELIT Software.
(An equivalent screen is available for the Sample Addition method.)
The ELIT software guides the user reliably through the measuring procedure. It calculates the
required concentration and volume of the standard from the volume and estimated concentration for the sample and checks the quality of the
results - i.e. if the estimate of the sample concentration was badly wrong and the ratio of sample to standard was not optimum then the operator is prompted to try again with a more suitable standard.
Note: The "Suggest" boxes are the calculated values suggested by the software but the "Actual" are the nearest more practical or convenient values which are typed in by the operator and used in the calculations.
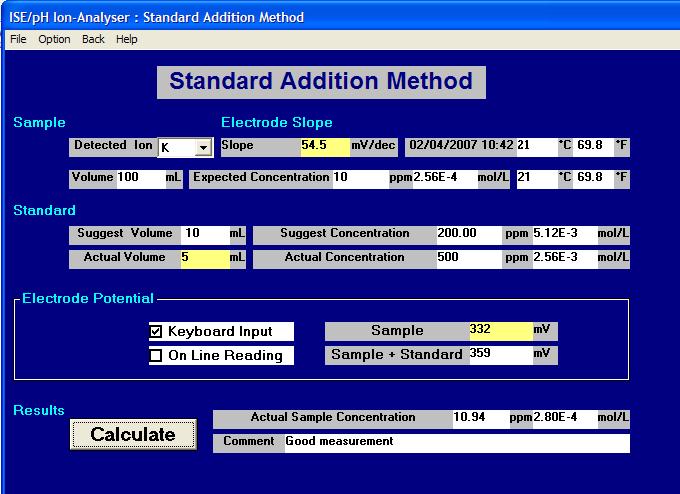
Introduction to Standard Addition and Sample Addition Methods.
CAUTIONARY NOTE: There is a common misunderstanding that these incremental methods will also help counteract the effects of Ionic Interference - but this is not so. If interfering ions are present in sufficient concentration, then they will always add to the signal produced by the primary ion and increase the measured voltage. The measuring system has no way of distinguishing between the signal produced by the primary ion and that from the interferent. Thus significant concentrations of interfering ions will always produce a falsley high reading no matter what method of analysis is used.
BASIC PRINCIPLE: These methods involve measuring the voltage in a relatively large volume (e.g. 25 - 100 ml) of sample (for Standard Addition) or standard (for Sample Addition) then adding a much smaller volume (e.g. 1 - 10 ml) of standard (or sample) and taking a second reading after the voltage has stabilised in the mixture. Note that this method will only work within the linear range of the electrode so cannot be used for low-level samples. The volume and concentration of the standard and the volume of sample must be chosen carefully according to the following criteria:
1) The volume of the first solution must be large enough to cover the tips of both electrodes and allow room for a magnetic stirring rod to continuously stir the solution during measurement and mixing of the additive - whilst ensuring the electrodes remain immersed.
2) The concentration and volume of the additive must be sufficient to cause a significant and measurable increase in the measured voltage of the first solution (ideally, 20 to 30 mV - achieved by increasing the original concentration by 5 to 10 times).
3) The volume of the additive must be small enough so that it does not cause a significant change in the Ionic Strength of the first solution.
4) The volume of the additive must be large enough so that volumetric errors are not significant - or use Gravimetric Addition - i.e add standard from a weighed "dropping bottle" then re-weigh.
QUICK GUIDE to Standard Addition Measurements using ELIT ISE/pH Ion Analyser Software
NB: Full instructions can be found in the Software Manual
1) First calibrate the electrodes in the conventional manner to find the slope in the range of the samples to be measured.
2) Determine the volume and concentration of standard, and volume of sample required by entering values into the on-line table.
3) Pippette the desired volume of sample into a measuring beaker and measure the voltage between the ISE and Reference electrode in the appropriate way for the ion to be measured (ie with or without stirring, sufficient time for stable reading).
4) Add the appropriate volume of standard, mix well and remeasure the voltage as before.
5) The Software will then calculate the concentration of the sample and give an assessment of whether the Sample to Standard ratio was optimum for this sample.
Summary of Advantages over Direct Potentiometry
- The electrodes remain immersed throughout the process so that there is little change in the liquid junction potential of the reference electrode (which can often be changed by several millivolts when the electrodes are removed from one solution and placed in another) between calibration and sample measurement - and therefore this source of measurement error is virtually eliminated.
- Calibration and sample measurement are both made essentially at the same time and in the same solution so that ionic strength and temperature differences between standard and sample are reduced and ISAB is not generally required.
- Once the approximate concentration for the samples is known, the calibration (slope) can be "fine tuned" by analysing a standard with a concentration that lies within the range of the samples (and is at the same temperature) and then adjusting the slope and re-calculating the results until the standard gives the correct answer. This "fine tune" procedure is very quick and easy using the ELIT ISE/pH Ion Analyser Software.
- Measuring the slope at or very near to the sample concentration means that these methods can be used with old or worn electrodes which may not be completely linear over their whole range, as long as the slope is stable and reproducible over the limited range of the samples.
See: www.nico2000.net/datasheets/StndAddnStats.xls
and www.nico2000.net/datasheets/Sampaddnstats.xls
The main disadvantage of these methods is that it is necessary to mix together accurately measured volumes of standard and sample, and they involve more complex calculations than simple direct potentiometry. This makes them a more skilled and time-consuming procedure and they have traditionally been less popular with most ISE users. It must be noted, however, that these analyses can now be made much more easily and quickly using the ELIT ISE/pH Ion Analyser (2-channel computer interface) software to calculate a suitable volume and concentration for the standard, measure the electrode potentials, and calculate the results - and by using automatic syringe pipettes to dispense the measured volumes.
Further slight disadvantages are that thes calculations are only valid within the linear range of the electrode and hence these methods cannot be used for low-concentration samples, near to the detection limit, and the approximate concentration of the sample must be known before commencing the analysis in order to choose an appropriate standard concentration and suitable volumes for the two solutions. Nevertheless, if the sample concentration is completely unknown, it can easily be determined by making a quick Direct Potentiometry measurement first. This can be done using an old calibration graph - but the most accurate results can only be obtained if the electrodes are calibrated using two standards which span the expected range of the samples immediately prior to analysis.
The Standard Addition (also known as "Known Addition") method is the most common and involves adding a small volume of a concentrated standard (e.g. 2, 5 or 10 ml) to a much larger volume of sample (e.g. 25, 50 or 100 ml). The voltage is first measured in the pristine sample. Then the standard is added, the solutions are mixed well, a second reading is taken and the software calculates the concentration of the sample. The main limitation to the Standard Addition method is that it requires relatively large volumes of sample (at least 20ml, preferably 100).
The Sample Addition method is used in exactly the same way as Standard Addition, except that a small volume of sample is added to a larger volume of standard. This method is most useful when only small amounts of sample are available, or for samples with relatively high concentration (generally needs to be above about 100ppm for method to work well), or those with a complex matrix. In the latter case it is important to achieve a sufficient dilution of the sample matrix so that it does not significantly alter the ionic strength of the standard and it may be beneficial to dilute the sample first before adding it to the standard; but care must be taken to ensure that the concentration of the measured ion is still sufficient to cause a detectable change in the mV reading in the pure standard.
Basis of Calculations
These methods depend on the fact that the plot of electrode potential (mV) against concentration (ppm or mol/L) is a logarithmic curve. Thus any particular ratio of the amount of increase in mV in response to a particular increase in concentration (i.e. the slope of the curve) will only fit in one unique part of the curve.
The formulae for the Standard Addition calculation is:
Where,
And for Sample Addition:
Alternative procedures for highest possible precision.
Partly based on ideas published by Denis Rice, Consulting Analytical Chemist,
64 Cliff Drive, Katoomba, NSW 2780, Australia. tdrice3@southernphone.com.au
" Gravimetric Analyte Addition Ion-Selective Electrode Potentiometry with Initial Spiking"
ISBN 0-646-37757-4.
This measures the calibration slope and the y-intercept and the sample concentration in quick succession whilst the electrodes remain immersed in the same solution. This eliminates any errors due to variation in Liquid Junction Potentials (mainly of the Reference Electrode) when electrodes are transferred from one solution to another and reduces errors due to differences in temperature or Ionic Strength between sample and standard solutions. Also the slope is measured in the correct concentration range for each sample.
This is potentially the most precise and accurate way of using ISEs but is rather more complex, analytically demanding and time-consuming. Also there is no Sample Addition equivalent so it cannot be used for high concentration samples. However, these can be diluted first to come within the useful range for the method (preferably below about 100ppm, but above the lower limit for linearity).
Click Here For full details of the method and an MS "Excel" spreadsheet to calculate the results.
Other potential refinements for the Single Standard Addition are as follows:
Adding ISAB to samples and standards will usually reduce the time needed for a stable reading and reduce even further any errors due to differences in Ionic Strength between standard and samples.
Always measure the electrode slope by Standard Addition to a known Standard solution immediately before measuring a batch of samples. The standard solution should have the same temperature as the samples and a concentration which is within the range expected for the samples. This procedure will minimise any error due to changes in the liquid junction potential of the reference electrode, "ageing" or non-linearity of the ISE, and variations in temperature during the analytical session.
The Slope is calculated as follows:
- m = (E2-E1) / [(Log C3) - (Log C1)]
- Gravimetric Addition
Add the standard solution from a weighed dropping bottle until the voltage has increased by between 20 and 30 mV. Then re-weigh the bottle - this will give a more controllable increase in the voltage, and the amount of standard added can be measured more precisely by gravimetric addition than by volumetric addition.
Gravimetric addition will also be more accurate for measuring small volumes and thus be helpful when only small samples are available and also allow greater flexibility in choice of volumes to use.
For even greater accuracy, weigh a known volume of standard to find its density in g/ml and divide the weight of standard added by this to get the accurate volume.
- Initial "Spiking"
For samples with concentrations which lie between the lower limit of linearity and the detection limit of the electrode, the sample must first be "spiked" with a sufficient quantity of the target ion to ensure that the measurements will be within the linear range. Then the sample is analysed in the conventional way by the Standard Addition method and the initial concentration is found by subtracting the initial "spike" from the final result.
This procedure is also useful for reducing the time needed to obtain a stable mV reading (which normally increases considerably as sample concentration decreases) - but it may only be effective for improving the precision of measurements for a very limited range of samples - probably only down to about 1 tenth of the lower limit of linearity.
For example, in the best case scenario, the overall error on ppm is probably not less than ±2% RSD (4% at 95% Confidence Level !) Thus if the lower limit of linearity is 1ppm and any sample below this is "spiked" to bring it up to, say, 2ppm, then the error on the spiked sample will be at least ±0.04ppm. So, after subtracting the spike, any sample with less than 0.1ppm will have an error of greater than ± 40% RSD !
- Compensate for Temperature Changes
If working in an environment where solution temperatures are likely to change by more than 2°C between measuring the slope and measuring the sample then it may be worth re-calculating the slope at the sample temperature before using it in the sample calculation. This simply requires the measured slope to be divided by the standard solution temperature - in Absolute or Kelvin degrees (0°C = 273°K) and multiplied by the sample temperature.
This procedure will save time by avoiding the necessity to make a new slope measurement every time the temperature changes, but will not be as effective because, in addition to affecting the electrode response to the target ion, temperature changes also affect the solubility of salts in the electrolytes and the development of junction potentials at the various metal-metal, liquid-metal and liquid-liquid interfaces in the system.Last Updated by CCR, 12 May 2017
and C3 = [(C1 x V1) + (C2 x V2)] / (V1 + V2)
Where:
C1 & V1 are the concentration and volume of the initial standard,
C2 & V2 are the concentration and volume of the added standard
C3 = the concentration of the mixture after adding the standard.