Innovative technology at highly competitive prices.
|
|
ELIT Brand Electrochemical Sensors and Computer-based Instrumentation. Innovative technology at highly competitive prices. |
| HOME | PRODUCTS | TECHNICAL INFORMATION | SITE MAP | CONTACT US | PRICE LIST |
|
Click here
to download a printer-friendly (.pdf) version of this document (11xA4 pages). INTRODUCTION ELIT solid state ion selective electrodes are fitted with either impregnated PVC or crystalline membranes, depending on which ion is to be detected. They are all mono electrodes which must be used with an appropriate reference electrode and must be inserted into an appropriate ELIT electrode head for connection to the measuring system. A)
A Quick Guide to Basic Operation
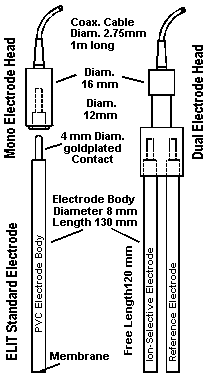
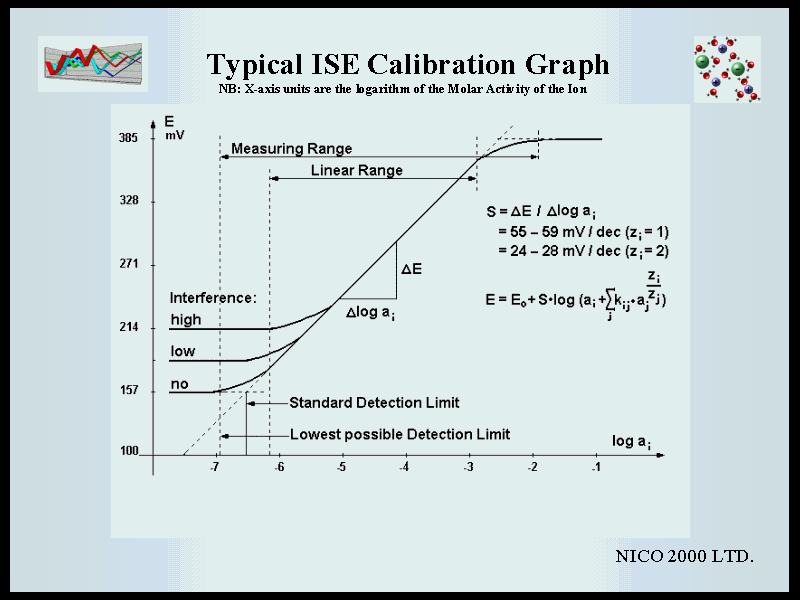
B) Detailed Instructions with Explanations and Discussion B1) Installation If the electrode is to be used with a conventional reference electrode, it must be inserted in a Mono Electrode Head (ELIT 101). This head fits in every standard electrode holder and is attached to a cable fitted with a BNC connector which can be connected to any ELIT Ion Analyser (computer interface), or to any pH/mV or ion meter with a BNC socket. Alternative DIN, US, or S7 connectors can be supplied if required. If the electrode is to be used in combination with an ELIT 8mm reference electrode, then both must be inserted into a Dual Electrode Head (ELIT 201). This system replaces conventional Combination Electrodes and has the advantage that the expensive low noise connectors and cables are not attached directly to the electrodes, the ISE or reference electrode can easily be exchanged independently and the electrode head can be re-used many times with any ELIT electrode. The black plastic protective caps must be removed from the electrodes before use. It is recommended that this is done before inserting the electrode into the electrode holder to avoid placing undue stress on the connecting pin. In order to obtain the most stable results, it is recommended that the electrodes are pre-conditioned by immersing both the ISE and the reference electrode in the 1000ppm standard for at least 10 minutes before use. If the voltage is monitored during this period and is seen to settle down to a sensible stable reading then this is a good indication that the system is operating correctly. Optimum wetting and pre-conditioning can be ensured by repeatedly removing the electrodes from the solution, washing and drying, and re-immersing (and waiting for at least two minutes for a stable reading) until three consecutive readings are within 1mV of each other. When the electrode is first used, or after prolonged storage, it may be that this procedure is insufficient and further soaking, overnight or over a weekend, may be necessary to achieve stable readings. In this case it is strongly recommended that the reference electrode is removed and the protective cap (containing outer filling solution) is replaced during this time to avoid unnecessary leaching of the filling solution. After preconditioning, rinse with a jet of de-ionised water and gently pat dry with a low-lint tissue. Preconditioning/Standard Solution B3) Calibration Theory.The operation of ion selective electrodes is based on the fact that there is a linear relationship between the electrical potential developed between an ISE and a reference electrode immersed in the same solution, and the Logarithm of the activity (or "effective concentration") of the ions in the solution. This relationship is described by the Nernst equation:
Note that 2.303RT/nF is the Slope of the line and this is an important diagnostic characteristic of the electrode - generally the slope gets lower as the electrode gets old or contaminated, and the lower the slope the higher the errors on the sample measurements - eg: at S=55, a 1mV error in reading will make about a 4% difference in concentration; at S=26 the difference will be more like 8%. Because of the logarithmic relationship, the slope can most easily be determined as the difference between the voltages measured in two solutions which differ by one order of magnitude - usually expressed as mV/decade. The theoretical value for the slope at 25°C is 59.2 for mono-valent ions and 29.6 for di-valent ions - but in practice these can vary considerably due to failure to meet "ideal" conditions. The critical factor is not so much the actual value of the slope but that this should be as high as possible and remain constant over the range of concentrations and the time period required for the analyses.
The activity of an ion in solution is a measure of the number of ions taking part in any given reaction, in this case those interacting with the ISE membrane. It is always less than the actual number of ions present in the solution (i.e. concentration) because the mobility of the ions is reduced by the presence of other ions in the solution. The higher the concentration of other ions, whether the same or different from the species being measured, the stronger is this retarding effect and the greater the difference between activity and concentration. However, it must be noted that in dilute solutions with low Ionic Strength this difference is small and can be ignored in many practical applications – i.e. the calibration can be made and sample results calculated using the more convenient concentration units. The relationship between activity and concentration is defined by the activity coefficient The Ionic Strength (I) can be calculated from I = 0.5 x Sum (ci x Zi2) Note that it is generally accepted that this formula is only accurate up to about I = 0.1 Molar. At higher ionic strength other factors come into play which make the calculation of activity coefficients virtually impossible and thus most ISEs cannot be used reliably above this concentration. Table 1 can be used as a guide in deciding whether or not the samples to be measured have sufficiently low Ionic Strength that the activity effect can be ignored - but also see Notes on Measuring Procedures (section B5,3). Table 1. Activity Coefficients and likely errors (i.e. under-estimate) in concentration measurement for various Ions in different Ionic Strength Solutions.
1) Bring the ionic strength to the same level in both the calibrating standard solutions and the samples by adding a suitable Ionic Strength Adjustment Buffer (ISAB) to both. 2) Dilute the samples to a level where the ionic strength effect is insignificant – but make sure that the detected ion is still within the linear range of the electrode. 3) For samples with complex but known matrix, make up the standards in a similar solution which does not contain the detected ion. 4) Use the Activity Coefficient to calculate the concentration from the activity. The activity coefficient can be calculated for simple solutions with known concentrations of all the ions, but this is not possible in many practical applications, where the samples may have a complex or unknown matrix. NB: the conversion between activity and concentration can be done automatically using the ELIT Chemtools software available with the ISE/pH Ion Analyser. Also see: Ionic Strength and activity calculator (MS Excel file) 5) Use the Standard Addition (or Sample Addition) Method where the voltage is measured before and after a measured small volume of standard (sample) is added to a larger measured volume of sample (standard) so that the ionic strength is essentially the same for both the calibration and the unknown measurement. These methods also have the added advantage that the electrodes remain immersed in the same solution for both measurements, thus minimising any errors due to temperature differences or hysteresis or variations in the liquid junction potential of the reference electrode, and thus can potentially yield more precise results than direct potentiometry; even for low-ionic-strength samples (click for details). B4) Calibrating the ElectrodeFor a complete calibration, a 10,000 or 1,000 ppm standard solution (or 0.1 or 0.01 molar solution, whatever is most convenient) can be diluted sequentially to produce concentrations of, for example, 100ppm, 10ppm & 1ppm or 1, 0.1 & 0.01 mMol (or even lower for some electrodes – see electrode specification sheet). Note that these are concentrations of the ion to be measured, not the salt containing the ion. The easiest way to do this is to pipette 10mls of the higher concentration solution into a 100ml flask, dilute to 100ml with deionized water and mix thoroughly. Then transfer this solution into a 150 ml polypropylene beaker ready for analysis. Then wash and re-use the pipette and flask, taking the next aliquot from the previous beaker. However, if the approximate concentration range of the samples is known then a complete calibration will not be necessary and standards should be made which closely bracket the sample range. Note that large errors may occur if samples are measured by extrapolation beyond the range of the calibration and it is easiest to calculate the slope, and check the performance of the electrode, if decades of concentration are used, e.g. 10 & 100 ppm or 1 & 10 mmol etc. - because the slope is simply the difference between the millivolts in two solutions which differ by one order of magnitude. Three or more calibration point are recommended in order to confirm the linearity and to detect any errors in diluting the standards, or to define the curve in the non-linear range. Once the slope has been confirmed in the range of the sample concentrations, subsequent re-calibrations can be made more quickly with a single standard in the middle of the sample range (at least on the same day and probably over a longer period for less precise measurements). This is relatively easy using the ELIT ISE/pH Ion Analyser software. The temperature of the calibrating solutions and the samples should be the same within a tolerance of ±1°C. If ISAB is required then this must be added in equal proportions to all standards and samples. Note that if the electrode specification sheet indicates that ISAB should be added, for example, "2% v/v" then it should be realised that these are only approximate proportions and the critical factor is that all solutions, samples and standards, must be treated in the same way. Thus it is generally easiest to add 2ml of ISAB to 90ml of standard solution (remaining after serial dilution), after the appropriate concentration standards have been made. In this case, 10ml must be removed from the lowest standard to make it the same as the others, and only 90mls of sample should be used. When the calibrating standard solutions are ready they should be measured in the same way as the samples (see Notes on Measuring Procedures, below) - working from the lowest concentration standard to the highest in order to minimise cross contamination. The mV reading must then be plotted against the log of the concentration (or against the concentration on a semi-log scale) on graph paper or computer graphics package. This is done automatically if an ELIT ISE/pH Ion Analyser (2-channel computer interface) is used instead of a conventional mV/ion meter. The mV reading for the samples can then be used to read the concentration of the samples from the calibration graph. Again, this is done automatically by the ELIT software. For the most precise results it is best to measure samples soon after calibration; ideally, calibrate before every sample. This can be done initially with two standards to define the slope, then, to save time, with a single standard using the same slope. In practice, the operator must decide what is the best compromise between the increased time needed for frequent re-calibrations and the precision required for his measurements. For many applications, calibrating once every hour or every 20 samples should be sufficient - but much less frequently if monitoring long term changes in a single solution. B5) Notes on Measuring Procedures The basic method of measuring standards and samples is as given in the Quick Guide (above). Nevertheless, for the most precise results, the operator should be aware of a number of factors and alternative methods which may affect the quality of the analyses. Thus, if the highest possible precision is required, it is recommended that, before starting any new type of sample, the operator should test the various alternative methods to see which will give the most reproducible results with his particular samples and electrode system. 1) Stirring the solution during measurement ? 2) How long to wait after immersion, before taking a reading ? Alternatively, it may be more convenient (and quicker) to take all readings after a pre-specified time after immersion. Normally the measured voltage changes fairly rapidly and falls by several millivolts during the first 30 seconds after immersion but then the change gradually gets slower and slower. Taking a reading after 2 - 5 minutes (depending on electrode system) will ensure that the initial equilibration of the ISE is completed and the reading is taken in the shallow part of the curve where the final stabilisation of the reference electrode liquid junction potential is only making small changes to the measured voltage. A third alternative is to observe the drift in reading as the electrodes equilibrate after immersion and then take a reading at the point where the direction of drift is definitely reversed - i.e. a different electrochemical process begins to dominate. 3) To use ISAB or not ? 4) Cleaning between samples ? C) Standard Addition and Sample Addition Methods.
The Standard Addition method (also known as "Known Addition") involves adding a small volume of a concentrated standard (e.g. 2 or 5 ml) to a much larger volume of sample (e.g. 50 or 100 ml). The volume and concentration of the standard must be chosen to cause a significant and measurable change in the concentration of the detected ion (and hence in the measured voltage) but should not cause a significant dilution of the sample matrix (so that the ionic strength remains essentially unchanged). The voltage is first measured in a measured volume of the pristine sample. Then a measured volume of standard is added, the solutions are mixed well and a second reading is taken before calculating the concentration of the sample. The Sample Addition method is used in exactly the same way as Standard Addition, except that a small volume of sample is added to a larger volume of standard. D) Interfering Ions The biggest limitation and difficulty with Ion Selective Electrode measurements is the problem of interference from other ions. ISEs are not ion-specific. All are sensitive to some other ions to some extent. In many cases the interferences are trivial and can be ignored but in some extreme cases the electrode is far more sensitive to the interfering ion than to the primary ion and can only be used if the interfering ion is absent, or only present in very low concentrations relative to the primary ion. In some systems the interfering ion can be removed by chemical means and special chemicals can sometimes be added to the appropriate Ionic Strength Adjustment Buffer. The interfering ions and their Selectivity Coefficients (SC=1 if electrode is equally sensitive to both the primary and interfering ions, 0.1 if ten times less sensitive to the interfering ion, etc.) are clearly shown on the individual electrode data sheets. Unfortunately the Selectivity Coefficient is not constant and is dependent on a number of factors such as total ionic strength of the solution, temperature and the actual and relative concentrations of each ion. Thus it cannot be used to make precise corrections for the interference. Nevertheless, the Chemtools package in the ELIT ISE/pH Ion Analyser software can be used to calculate the order of magnitude of the likely interference effects for all ELIT ISEs as a guide for the user in deciding whether or not ISE measurement would be suitable for his application. If the analyst is unsure if an interfering ion will be a problem, or if he wishes to make a correction for its presence, then it is possible to measure the SC directly in a typical sample and obtain a more accurate assessment of the interference. First measure the concentrations of the primary ion and the interferent in the sample, using the appropriate ISE for each ion. Then add more interferent - sufficient to ensure a significant (say 10%) increase in the signal for the primary ion. The amount to add can be calculated from the initial concentration measurements and the indicative SC quoted in the electrode specifications. Then measure the apparent concentration of the primary ion again and calculate the SC for the interferent from the increase caused by the known amount added. e.g. if the added interferent increased it's concentration by 10ppm and this produced a 1 ppm increase in the measured value of the primary ion then the SC would be 0.1. Subsequent sample measurements can then be made by measuring both the primary ion and interferent and using the measured SC to make a more reasonable correction for the interference. Click here for practical example. These measurements can be made by Direct Potentiometry or Standard Addition, with or without ISAB, depending on whichever method has been chosen for the samples. However, it must be noted that the accuracy and precision of this correction may be quite variable and will need to be tested for validity and reliability for each particular application before being used on a routine basis. E) Storage and Maintenance After use, the electrodes should be rinsed with de-ionised water and dried with a paper towel. If the electrodes are in regular use and are kept permanently plugged into the electrode head, then it is not necessary to replace the plastic cap for the ISE after every analytical session. It can just be left hanging in the air. If unused for long periods however, (overnight or for prolonged storage ) it is necessary to replace the cap to protect the membrane from atmospheric oxidation/corrosion or mechanical damage.
The cap for the reference electrode however, should be replaced after each session of use, in order to prevent drying out of the filling solution. Reference electrodes should be stored wet, with the protective cap containing the same solution as the outer filling solution. Loss of solution is minimised by keeping a plug of absorbent material, such as cotton wool, in the bottom of the cap. This should be checked frequently and the solution replenished if necessary. NOTE 1: Electrodes should never be left in pure distilled or de-ionised water (or any solution which does not contain the target ion) for more than a few minutes at a time - to avoid leaching effects. F) Fault Finding Important Diagnostic Note: It is quite normal for the measured voltage in a particular concentration standard to differ from a previous calibration (on the same day or days or weeks before) or from another ISE for the same ion. This does not necessarily indicate a fault. (1) If the electrode shows a slow response or a low slope, the membrane may be poisoned or coated with a film. In this case PVC membranes may be rejuvenated by carefully washing in pure ethanol, then de-ionised water. Thick deposits can be washed off with a jet of deionised water from a wash bottle. As a last resort, try gentle wiping with a soft wet tissue or lint-free cloth - but be very careful not to scratch the membrane. Protein deposits can be removed by rinsing in a HCl/Pepsin cleaning solution available from several chemical suppliers. (2) If the ISE is being used with an ELIT 8mm reference electrode and the signal is very erratic and jumps by tens or even hundreds of millivolts then this is probably due to minute bubbles in the reference electrode gel electrolyte. These can develop during transport or prolonged storage. This can normally be cured by holding the reference electrode firmly with the active tip pointing downwards and shaking down several times with a flick of the wrist, as with old medical mercury thermometers ( i.e. down vigorously but up gently so that the gel is propelled towards the ceramic frit and any bubbles away from it). (3) If the reading when immersed in the 1000ppm standard is 0 mV or an unchanging high value then it is likely that the gel electrolyte in the reference electrode has completely dried out. This may be because there has been insufficient filling solution in the plastic cap or the cap has not been replaced after use. In this case a new RE will be required. (4) Large deviations in the mV reading may also be due to poor connections in the wiring or moisture on the contacts and in this case all connections should be checked, cleaned and dried. Random deviations of a few millivolts may be due to contamination of the ISE membrane or to external electrostatic fields. In the latter case, if the operator or passers-by are wearing static-producing clothing, it may help to ensure that every one remains still for a few seconds whilst the reading is taken. |